Группа российских ученых, среди которых сотрудники Московского физико-технического института, предложила новый метод сравнения метагеномов — совокупности последовательностей ДНК всех организмов в образце исследуемого биологического материала. Метод позволяет более эффективно и быстро решать задачу сравнения образцов и может быть легко внедрен в процесс анализа данных в любом метагеномном исследовании. Работа опубликована в журнале BMC Bioinformatics.
Среди объектов исследования метагеномики особое место занимают бактерии, населяющие организм человека. Значимость метагеномики нельзя недооценивать: по примерным оценкам, в нашем организме на порядок больше бактериальных клеток, чем наших собственных, причем большая часть из них находится в кишечнике. В ходе различных глобальных проектов, таких как «Микробиом человека», было выявлено, что состав бактериального сообщества влияет на риск возникновения заболеваний, выбор оптимального режима питания, настроение и даже творческие способности. Также и наоборот — состав микроорганизмов чутко реагирует на процессы, происходящие в организме. Таким образом, путём сравнения образца пациента с кишечными метагеномами здоровых людей можно в перспективе оценить риск опасных заболеваний, таких как сахарный диабет или воспалительные заболевания кишечника.

Бактерии микрофлоры кишечника. (Источник изображения: http://laktiale.com.ua/uploaded/Microflora.gif )
Традиционным подходом в метагеномном анализе является сравнение образцов на основе их таксономического состава — процентных долей каждого найденного микробного вида. Для того, чтобы определить состав образца, его последовательности сопоставляют базе известных бактериальных геномов, называемых референсным набором. Однако такой подход имеет ряд недостатков. Во-первых, референсные геномы зачастую неточны, поскольку составление референсного генома — вычислительно сложная и трудоемкая задача, особенно для труднокультивируемых видов организмов; а геномы изолированного в лаборатории вида могут нести набор генов, существенно отличающийся от того же вида, обитающего в естественной среде. Во-вторых, не для всех организмов в принципе существуют собранные референсные геномы; примерами таких организмов являются вирусы. Поэтому та часть последовательностей образца, для которой не найдено соответствие с референсом, просто не учитывается в процессе анализа, несмотря на то, что она может быть достаточно объемной и значимой. Между тем, метод, основанный на сопоставлении частот k-меров, не требует обращения к референсу и наличия какой-либо информации об исследуемых организмах, и поэтому анализу подвергаются уже все последовательности образца, что дает лучшие результаты.
В основе метода лежит представление о последовательности генома организма как о наборе всех встречающихся в нем нуклеотидных «слов», заданной длины k, называемых k-мерами. Поскольку геном является уникальной для каждого организма последовательностью, то и наборы таких «слов» различаются между отдельными организмами. Таким образом, набор всех k-меров метагенома можно рассматривать как совокупность наборов, входящих в его состав организмов. Это позволяет судить о различиях в бактериальном составе при сравнении образцов между собой.
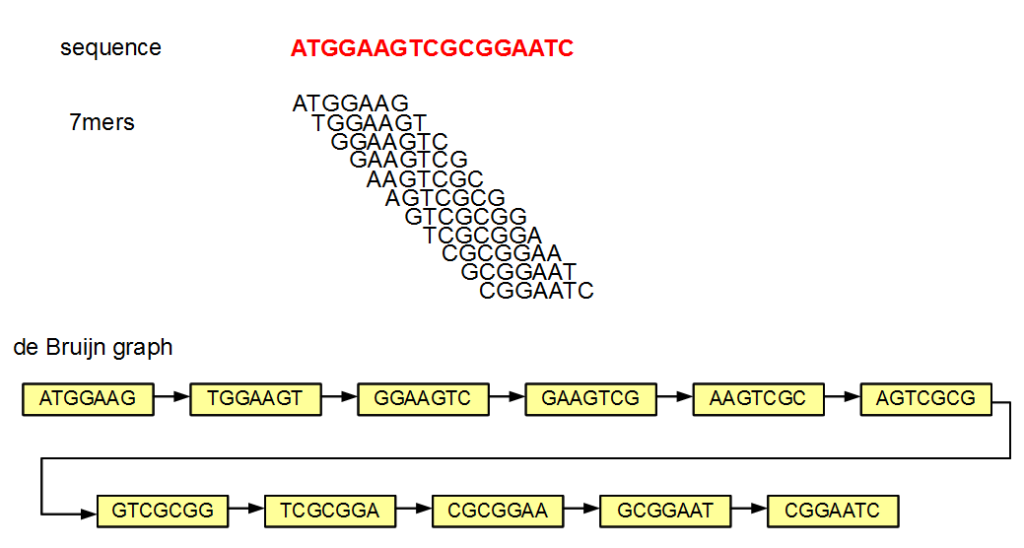
Пример выделения всех k-меров длины для k=7 из произвольной последовательности. (Источник изображения: http://www.homolog.us/blogs/wp-content/uploads/2012/06/i6.png)
По словам автора статьи, заместителя заведующего Лабораторией системной биологии МФТИ Дмитрия Алексеева: «Важно, что гены можно рассматривать не только как участки ДНК с закодированными в них белками, но и как просто информацию. Именно такое информационное различие позволило нам выявить новые участки ДНК — не описанные в каталоге известных генов. Интересно посмотреть, как такой подход будет использоваться другими группами».
Разработанная методика позволит более эффективно и точно находить отличия между метагеномами разнообразных бактериальных сообществ, что в частности может помочь в изучении, диагностике и лечении многих заболеваний человека.
