Международная группа ученых сделала большое обновление HOCOMOCO — базы моделей нуклеотидных последовательностей участков ДНК, связывающих транскрипционные факторы, созданной в 2013 году. Статья опубликована в журнале Nucleic Acid Research.
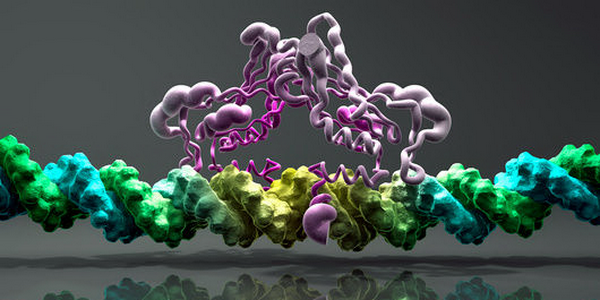
Модель взаимодействия транскрипционного фактора Brachyury с ДНК. Иллюстрация Phantatomix.
HOCOMOCO в переводе на русский расшифровывается как «Обширная коллекция моделей для Homo Sapiens». В этой базе хранятся модели участков связывания транскрипционных факторов. Каждая такая модель — это математическое представление участков ДНК, с которыми может связываться транскрипционный фактор — один из белков, которые подавляют или, наоборот, активируют работу различных генов. В геноме человека закодировано более полутора тысяч транскрипционных факторов. В базе данных хранятся модели участков связывания транскрипционных факторов для мышиных и человеческих ортологов — генов, которые произошли от одного и того же гена у вида — общего предка мыши и человека.
К такой базе, как HOCOMOCO, обращаются исследователи со всего мира для своих экспериментов. С помощью моделей из базы можно, например, предсказывать места связывания транскрипционных факторов с нуклеотидами в цепочке ДНК. То есть с такими местами в геноме, на экспрессию генов в которых как раз влияют транскрипционные факторы. После этого на основе предсказанных мест связывания можно строить модели регуляторных сетей, которые объясняют механизмы переключения генов в различных условиях. Такие сети нужны для понимания биологической картины экспрессии генов в том или ином процессе, например при развитии рака.
Для того чтобы создать такую базу, ученые собирали результаты экспериментов по исследованию взаимодействий ДНК и транскрипционных факторов из нескольких открытых баз данных. Большую роль в этой работе сыграли сотрудники ФИЦ информационных и вычислительных технологий Сибирского отделения РАН под руководством Федора Колпакова, заведующего лабораторией биоинформатики. Благодаря их работе удалось получить огромную коллекцию фрагментов ДНК, связывающихся с белками. Для создания HOCOMOCO в этих фрагментах с помощью вычислительного анализа ДНК-текста были найдены мотивы — небольшие последовательности ДНК, с которыми связываются транскрипционные факторы. Прежде чем мотивы попадут в итоговую базу данных, их аннотируют — с помощью специальных инструментов определяют структуру соответствующих белков и их функцию. Дальше для мотивов определяют надежность, показывающую, насколько достоверно взаимодействие несущей мотив ДНК и транскрипционного фактора в экспериментах разного вида.
Но это не единственная проверка. Перед попаданием в базу каждая модель проходит вычислительные эксперименты на то, насколько хорошо она помогает предсказывать места связывания ДНК с транскрипционным фактором. Результаты вычислительных экспериментов сравниваются с реальными данными, полученными в лаборатории. На основе нескольких разных сравнений каждой модели выставляются оценки точности, чувствительности и специфичности. После всех этих процедур составляется итоговая строчка для каждой модели в общую базу. База открыта, и ученые со всего мира могут использовать данные из нее для планирования своих экспериментов. По сравнению с предыдущей версией, в HOCOMOCO появилось больше новых моделей, возросла их точность и валидность. Кроме этого, построены коллекции специальных моделей для предсказания ДНК-белковых взаимодействий in vivo, in vitro, а также для предсказания индивидуальных вариантов в геноме, влияющих на связывание белков-регуляторов.
«Мы считаем, что HOCOMOCO — это надежная база данных, расширяющая возможности молекулярной биологии и эпигенетики. Для ее пополнения и обновления наша команда изучила данные 14 183 экспериментов ChIP-секвенирования и 2554 экспериментов HT-SELEX, что дало возможность получить более 400 тысяч мотивов-кандидатов, из которых было отобрано 1443 мотива, характеризующих участки ДНК, связывающие 949 транскрипционных факторов человека и 720 их аналогов у мыши», — говорит Всеволод Макеев, член-корреспондент РАН, заведующий лабораторией системной биологии и вычислительной генетики ИОГен им. Н. И. Вавилова РАН, заведующий кафедрой биоинформатики и системной биологии МФТИ.
Работа выполнена при поддержке Российского научного фонда (грант 20-74-10075).
В работе принимали участие ученые из Института общей генетики им. Н.И. Вавилова РАН, ФИЦ информационных и вычислительных технологий (Новосибирск), Института исследования белка РАН, МГУ им. М.В. Ломоносова, МФТИ, Института биохимии и генетики УФИЦ РАН, Сколтеха, Института проблем передачи информации РАН, НИТУ «Сириус», ООО «Биософт.Ру» (Новосибирск), НИЦ биотехнологии РАН, Казанского федерального университета, а также из США и Канады.
