
Нобелевскую премию по химии за 2024 год поделили создатели алгоритма AlphaFold — Демис Хассабис и Джон Джампер, — а также Дэвид Бейкер, автор алгоритмов из семейства Rosetta. Первые научили алгоритм машинного обучения предсказывать форму белка по последовательности его аминокислот. Алгоритмы, созданные вторым, помимо этого, также умеют решать обратную задачу: придумывать последовательность для искусственных белков нужной учёным формы.
Форма белка определяет его биологическую функцию. Молекулярный биолог, взглянув на белок, может, в целом, понять, выполняет ли тот какую-либо транспортную функцию, работает ли ферментом и даже то, с какими еще белками хорошо взаимодействует, а с какими — нет. Проблема, однако, в том, что для того чтобы посмотреть на белок, нужно проделать огромную работу.
У белка четыре уровня структуры.
- Первичная структура — это последовательность аминокислот, из которых состоит белок. Большинство белков на этом уровне представляют собой ту или иную последовательность, которую можно записать при помощи алфавита из 20 с небольшим «букв». Аминокислот намного больше двадцати, но большинство белков обходятся этим числом (почему это так, вопрос всё ещё открытый).
- Вторичная структура описывает то, какую форму принимают отдельные фрагменты цепочки аминокислот после того, как они были синтезированы на матрице РНК. Типичных форм тут тоже не так много: несколько видов спиралей, листы, «разное».
- Третичная структура — это финальная форма белка, которую тот принимает после укладки (фолдинга). И вот тут уже начинается астрономическое разнообразие фигур.
- Четвертичная уже описывает типы взаимодействий, в которые может вступать этот белок.
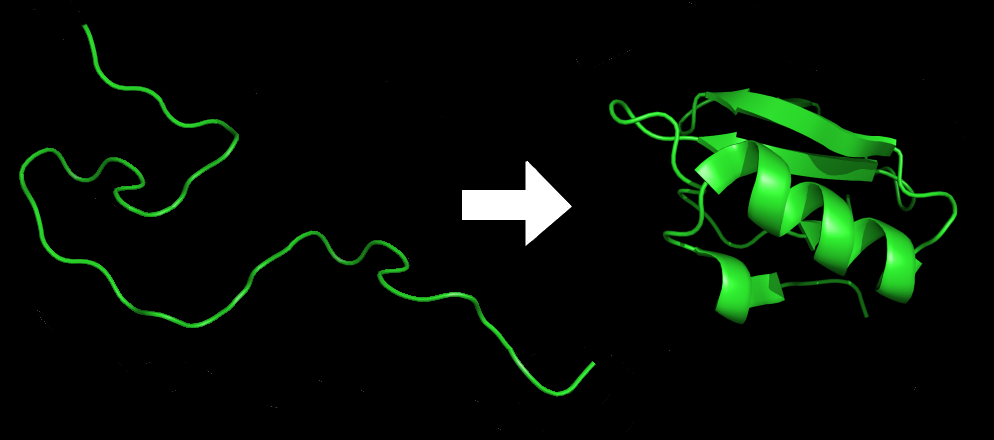
Цепь и третичная структура ингибитора химотрипсина
DrKjaergaard / wikimedia commons
Золотой стандарт определения третичной структуры белка — метод рентгеноструктурной кристаллографии. Для этого вам нужно аккуратно вырастить в лаборатории кристаллы интересующего вас белка, поместить их под рентгеновское излучение и собрать достаточное количество данных о том, как именно оно рассеивается на ваших кристаллах. Затем по ним вы можете восстановить изображение.
Этот метод был изобретен в 50-е годы прошлого века, его изобретатели получили свою Нобелевскую премию, и уже не одно поколение кристаллографов сделали себе научную карьеру, занимаясь портретированием белков для коллег. Само только описание третичной структуры иногда уже достаточное техническое достижение, чтобы претендовать на отдельную научную публикацию.
Это долго и сложно. Проблемы начинаются уже на самом первом этапе — получении кристаллов. В этом деле иногда участвуют космонавты, потому что в невесомости меньше рисков испортить форму особенно нежного образца. Но есть вещества, которые даже космонавты не могут заставить кристаллизоваться нормально. Есть и такие, что, например, меняют форму после кристаллизации.
Узнать первичную структуру намного проще, дешевле и быстрее. Так что в то время, как число белков, третичную структуру которых мы определили, до сих пор около 200 с небольшим тысяч, количество известных сиквенсов давно исчисляется миллионами.
Но по этим данным разобраться с функцией белка с достоверностью нельзя.
Но должно быть можно
В декабре 1972 года биохимик Кристиан Анфинсен приехал в Стокгольм, чтобы забрать свою половину Нобелевской премии. Часть протокола в таких случаях — noblesse oblige — прочесть перед собравшимися на торжественную церемонию лекцию. Анфинсен выступил с докладом об «Исследованиях принципов, которые управляют укладкой белковых цепей», где сформулировал термодинамическую гипотезу, также известную как догмат Анфинсена. Через полгода текст его выступления с некоторыми правками (в результате которых заглавие стало уже «Принципами, которые управляют укладкой…») опубликовал журнал Science, и эта статья по сей день остается классической.
Функция белка (а она выражается его формой), утверждал Анфинсен, — не случайна. Её суть уже зашифрована в последовательности аминокислот. Это подтверждают эксперименты по ренатурации белков. Свою премию Анфинсен получил именно за них. Его опыт выглядел так:
- подвергаем химической денатурации препарат рибонуклеазы;
- проверяем ферментативную активность денатурированных рибонуклеаз;
- убираем химические агенты и возвращаем препарат в исходное состояние;
- проверяем ферментативную активность рибонуклеаз, прошедших через эту процедуру.
После денатурации способность рибонуклеаз расщеплять нуклеиновые кислоты пропадала, а после ренатурации восстанавливалась (хоть и не целиком) — причём сама, in vitro, прямо в пробирке. Из этого Анфинсен выводил, что третичная структура белка заложена уже на уровне межатомных взаимодействий в полипептидной цепи.
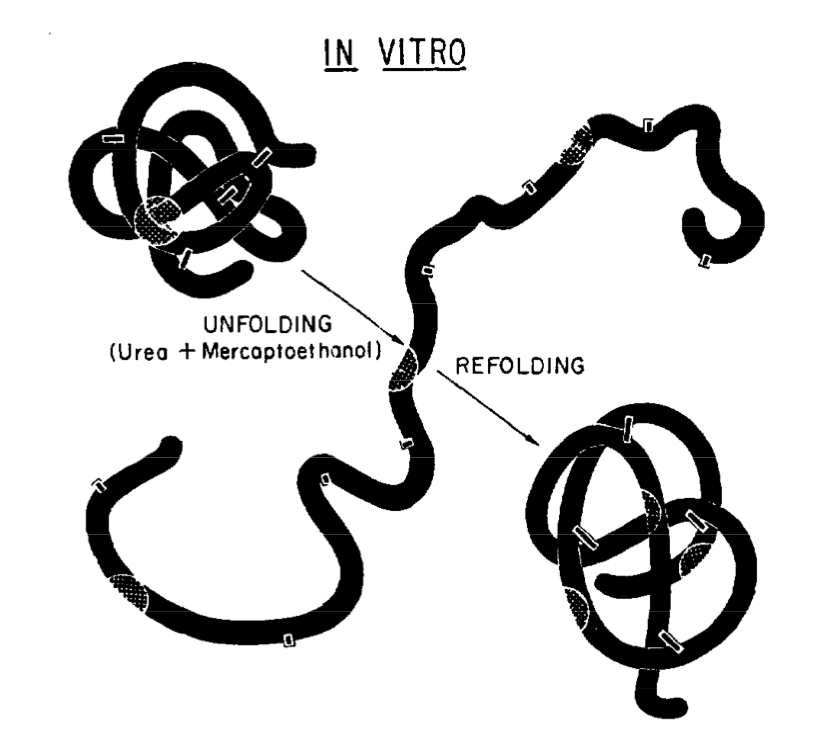
Схема эксперимента Анфинсена
Осталось только объяснить, почему это происходит — то есть вывести общие принципы, которые этим управляют. Их описание даст нам возможность предсказывать фенотип белка по его генотипу, равно как и заниматься рациональным дизайном новых белков.
Путь к этому прекрасному будущему, однако, упирался в проблему, которую сформулировал за несколько лет до этого Сайрус Левинталь (Cyrus Levintal). Дело в том, что у полипептидной цепи в свободном виде так много степеней свободы, что решение этой задачи перебором должно занимать время, сравнимое со временем существования Вселенной, даже если каждая итерация будет занимать у нее пикосекунду. При этом рибонуклеазы в экспериментах Анфинсена приходили в норму за несколько секунд.
Более того: уже упомянутые выше казусы, с которыми сталкиваются кристаллографы при подготовке образцов к исследованию, говорят нам, что нативная конформация белка не единственная возможная устойчивая форма его существования. А через 10 лет после выступления Анфинсена группа Стенли Прузинера сообщила, что им наконец-то удалось изолировать инфекционный агент, вызывающий у крупного рогатого скота губчатую энцефалопатию — и это белок. Причём не какой-то особый, а самый обычный мембранный белок млекопитающих PrP, просто он сложен в отличную от нормальной форму. И кроме того, что она гораздо более стабильная, чем «здоровая» — для его денатурации требуются намного более экстремальные условия — белки такой формы каким-то образом умудряются убедить своих соседей изменить форму. И это без участия нуклеиновых кислот или каких-либо ещё посредников. Отличия прионных белков от здоровых выражаются уже на уровне вторичной структуры: у прионного PrP доля бета-листов преобладает над долей альфа-спиралей, в то время как у «здорового» PrP всё наоборот.
Мы знаем, что происходит в ткани, в которой завелись прионные белки. Мы не понимаем, как и почему это происходит. Это просто происходит, и всё, что мы можем, смотря на происходящее — наблюдать. Летальность прионных болезней — стопроцентная.
С мира по нитке
В 1971 году журнал Nature разместил на одной из своих страниц объявление, что Брукхейвенская национальная лаборатория и кембриджская База кристаллографических данных создают Банк белковых данных (Protein DataBank, PDB). Система будет отвечать за хранение атомных координат, факторов структуры и карт электронной плотности белков, и делиться этими данными на магнитных лентах со всяким, кто обратиться к ней с соответствующих запросом. Услуги эти она будет предоставлять бесплатно. Успех предложенной системы будет зависеть готовности кристаллографов, занимающихся изучением белков, делиться своими данными — будь они «сырыми» или обработанными, в виде рукописей или машинными.
Вообще история PDB началась за три года до того. В 1968-м году завкафедрой химии Брукхейвенской национальной лаборатории Уолтер Хэмилтон (Walter Hamilton) понял, что изобретенный его коллегами-физиками инструмент BRAD (Brookhaven RAster Display) подойдет не только для того, чтобы рисовать объемные картинки треков распада элементарных частиц в пузырьковой камере, но и для изображения молекул.
Пример изображения, полученного при помощи BRAD
Хэмилтон пригласил своего знакомого Эдгара Мейера (Edgar F. Meyer) провести лето в Брукхейвене, чтобы воплотить эту идею в жизнь. Мейер до этого несколько лет проработал в MIT в группе Сайруса Левенталя (того самого, кто обратил внимание на парадоксальную скорость фолдинга белков) и уже имел необходимый для этого опыт программирования на Фортране.
Как вспоминает Мейер, он пропал для своей семьи на три месяца, потому что к компьютеру в Брукхейвене его подпускали после физиков, в третью смену — то есть между полуночью и рассветом. Итогом его трудов стала программа DISPLAY, которая занимала около двух тысяч перфокарт: она строила трехмерную модель молекулы, правда единовременно на экране не могло разместиться больше 512 атомов. Пользователь мог вращать, а также приближать и отдалять изображение, чтобы детальнее разглядеть интересующий его фрагмент.
Изображение миоглобина, которое получил в 1968-м году Эдгар Мейер
Год спустя Мейер снова вернулся в Брукхейвен, чтобы продолжить возиться с визуализацией белков. Из Европы Мейеру прислали «неофициальный» набор координат атомов миоглобина. Тот вручную перенес их на перфокарты и скормил машине.
В 1970-м Мейер выпросил у коллег данные о структуре еще двух белков — рибонуклеазы и субтилизина. В отличие от миоглобина, форму которого определили европейцы, эти структуры были получены в США, и для описания их вместо метрической системы использовалась имперская. После того, как Мейер конвертировал их вручную, он написал отдельную программу для того, чтобы впредь делать это автоматически. С этим ему помог Джон Коггинс, заодно подтянув знания программиста в области биохимии. Взамен Мейер научил Коггинса работать с машинкой для пробива перфокарт.
В то же лето Мейер с Хэмилтоном определились со стандартом записей в базе данных для визуализации структур: под каждый атом заводилась отдельная перфокарта, данные о его позиции кодировались при помощи пяти слов. Также их система оставляла место на каждой перфокарте для аннотаций — чтобы пользователи будущей библиотеки могли предупреждать коллег о возможных погрешностях в их данных или каких-нибудь особенно примечательных локациях.
Сделав всё это, отцы-основатели PDB занялись прозелитизмом — ездили по исследовательским центрам, рассказывали о своей работе и просили коллег принять участие в пополнении базы. Ключевой была встреча с людьми из Кембриджа, где в то время также собирали библиотеку кристаллографических данных. Договорившись об унификации формата записи, учёные поспешили сообщить о новой базе миру.
В 1971 году в базе PDB хранились данные о структуре семи белков. Через два года, в 1973-м, к ним добавилось еще два. Какой-то особенной пользы от них не было — в то время редкая лаборатория могла позволить себе купить всю необходимую технику, чтобы порадовать глаза своих сотрудников трехмерными изображениями на экране телевизора. Посылки из PDB, таким образом, для конечного пользователя выглядели как колонки цифр на внушительной стопке перфокарт.
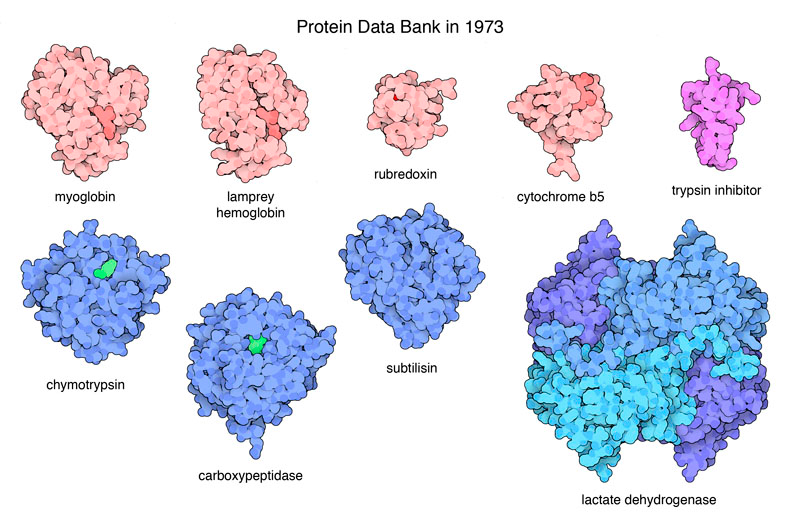
Изображения первых 9 белков, попавших в базу PDB
Но зачастую исследователя интересовал только конкретный фрагмент белка, а коробки с перфорированными карточками ставить ему было некуда. Поэтому Мейер написал программу SEARCH, которая позволяла ему после выведения на экран нужной картинки извлечь данные только о тех атомах, что прямо сейчас находились на экране. Либо извлекать из модели позиции только атомов конкретного типа.
В следующее лето Хэмилтон приехал к Мейеру в Техасский университет A&M — скорее навестить единомышленника, чем заниматься чем-то конкретным. Но его визит совершенно случайно пришелся на окончание внутреннего конфликта администрации университета с факультетом физики — теоретики постоянно занимали телефонную линию, потому что гоняли по ней данные на вычислительные машины в Остине и обратно, тратя на это неприемлемое, по мнению администраторов, количество денег. Услышав о том, что конфликт закончился победой бухгалтерии и канал связи скоро прикроют, создатели PDB решили под занавес организовать сеанс связи с Брукхейвеном по телетайпу. В результате они переслали себе данные о небольшом кусочке структуры миоглобина — их передача на три с лишним тысячи километров заняла полчаса. Так появился CRYSNET, а проект Хэмилтона и Мейера, наконец, обрел свою финальную форму.
Принципиальная схема проекта PDB
В 1999-м число записей в PDB стало уже десять тысяч. Остальные 215 с лишним тысяч попали в этот реестр уже в XXI веке.
В 2020-м году искусственная нейросеть глубокого обучения AlphaFold2, разработкой которой в недрах Google руководили Демис Хассабис и Джон Джампер, проглотила эти данные — и теперь предсказывает третичную структуру белков с точностью, иногда практически не отличающейся от экспериментальных методов. На главной странице одного из центров PDB рядом с общим числом структурных данных в базе появилось также число белковых форм, описанных «нейронкой» Хассабиса и Джампера. Счёт такой — 225 946 у команды экспериментаторов, 1 068 577 у алгоритма Google.
Но это не соревнование, конечно.
Из ниток — пряжа
В 1994 году Джон Молд (John Mould) и Кршиштов Фиделис (Krzysztof Fidelis) организовали конкурс CASP (Critical Assessment of protein Structure Prediction), чтобы стимулировать развитие вычислительных методов предсказания третичной структуры белка. С тех пор они договариваются с экспериментаторами, что те придержат публикацию некоторых своих работ и, соответственно, не будут добавлять новые записи в базу белковых структур PDB до определенной даты, к которой участники конкурса обязуются представить своё предсказание формы тех самых белков, которую уже определили кристаллографы. Конкурс проводится раз в два года.
Внутри самого конкурса есть задания разной уровни сложности. Например, какие-то белки предсказать проще, потому что они родственны уже известным. Самый сложный «снаряд» CASP, когда опереться модели вообще не на что, и решать задачу надо, отталкиваясь только от последовательности аминокислот.
Результаты участников оценивают по шкале GDT (global distance test) от 0 до 100. Она выражает то, насколько далеко отстоит модельная структура от реальных экспериментальных данных. С чисто прагматический точки зрения, решение задачи на уровне 90+ по этой шкале уже можно считать окончательным успехом, поскольку на такой дистанции уже невозможно нормально сказать, кто ближе к истине, экспериментатор или модель — всякое реальное измерение имеет погрешности.
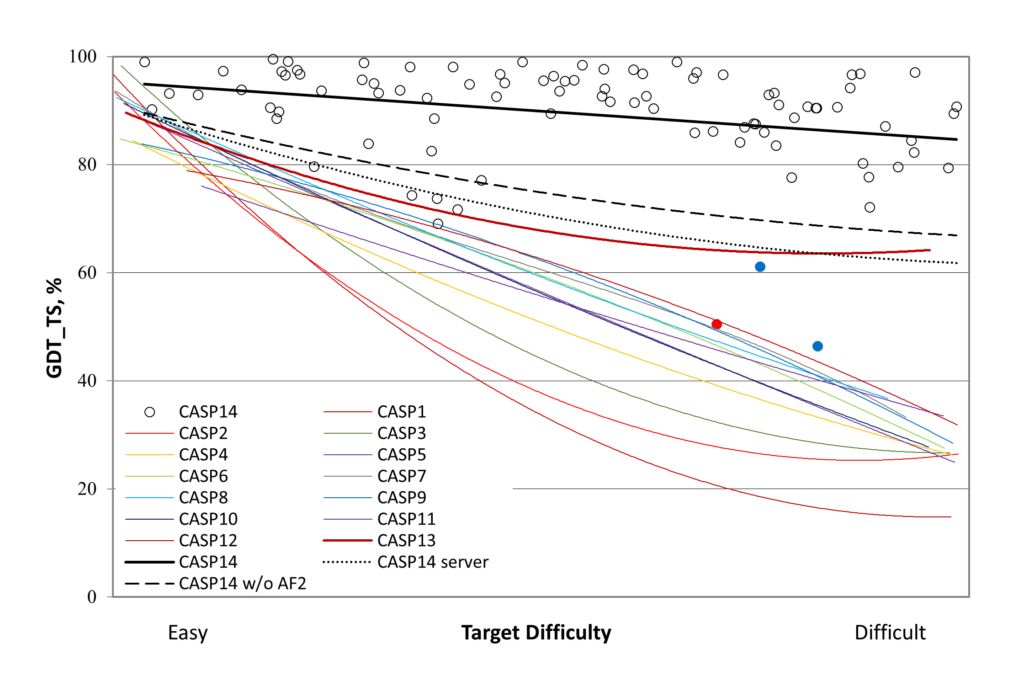
Успехи участников 14-ти конкурсов CASP (взятые в среднем) в предсказании третичной структуры белков разной сложности. Черной прямой — результаты участников CASP14 с учетом AlphaFold, пунктиром — без учета AlphaFold
Создатели CASP объясняют свою мотивацию, прямо ссылаясь на догмат Анфинсена — их интересуют принципы, которые руководят укладкой полипептидных цепей в белок. 30 лет назад более чем разумной казалась мысль, что для того, чтобы решить задачу, надо понять, как она решается. Вооружившись этим пониманием, ты можешь написать универсальный алгоритм решения интересующей тебя задачи.
В 1998 году Дэвид Бейкер с коллегами приняли участие в третьем конкурсе CASP — и с тех пор продолжали дорабатывать алгоритм Rosetta. В его базе — метод Монте-Карло, плюс, естественно, ряд авторских поправок и оптимизаций для того, чтобы повысить точность применительно к конкретной задаче предсказания третичной структуры. Но такой обсчёт все равно требует значительных вычислительных ресурсов, поэтому в 2005 году Бейкер запустил проект Rosetta@home. Это платформа распределённых вычислений, для которой нужны люди — точнее, их компьютеры. Любой мог — и может до сих пор — посодействовать делу решения фундаментальной проблемы: для этого надо просто поставить клиент Rosetta@home на свою машину и вместо того, чтобы выключать компьютер на то время, что он тебе не нужен, оставить его работать. Вскоре «Розетта» стала второй по размеру сетью распределенных вычислений в мире после биткойна — к ней присоединилось больше 200 тысяч человек.
Идея Rosetta@home, ровно как и идея самого конкурса CASP — развивает идеют, с которой наверняка могли бы солидаризироваться основатели PDB. Мы не понимаем, как цепочка аминокислот укладывается в тот или иной белок. Но исходим из того, что это временно, надо просто присмотреться, и понимание — придёт. То есть в какой-то момент количество изученных данных перейдёт в качество, и вот тогда-то настанет время кричать «эврика».
Это всё игры
Один из важных поворотов в истории Rosetta произошел, когда учёные поняли: их инструмент для предсказания третичной структуры белка можно применить и для решения обратной задачи. То есть сгенерировать аминокислотную последовательность, смотря на заданную форму. И эта форма не обязательно должна быть известна. В 2003 году Бейкер и его команда описали принципиально новый, искусственный белок Top7, последовательность которого складывалась в трёхмерную фигуру самостоятельно — хотя этот процесс выглядит не совсем типично, если сравнивать его с естественными белками.
Но главное для «нобелевской» истории с фолдингом белка произошло в 2008-м, когда команда Бейкера выпустила Foldit (буквально «Сложи это») — головоломку, игроки в которую соревнуются в складывании белков. Можно заниматься дизайном искусственных белков, можно попытать свои силы в предсказании третичной структуры по последовательности аминокислот (например, во время пандемии игроки возились со спайк-белком SARS-CoV-2), можно заняться сборкой малых молекул для лекарств.
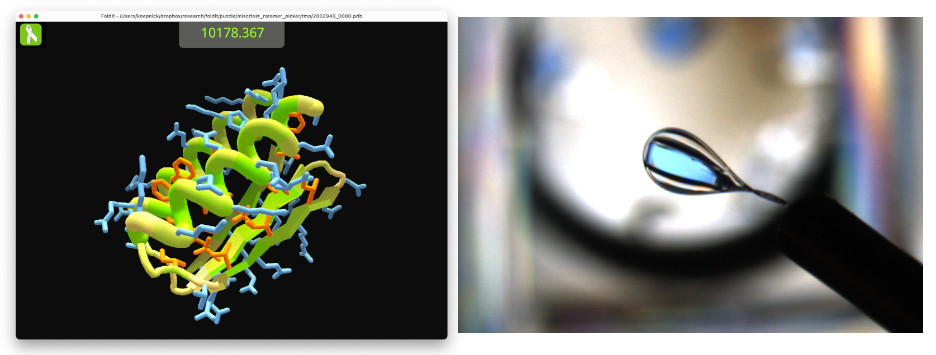
Слева: структура искусственного белка, созданного игроками в Foldit. Справа: мономерный кристалл этого же белка
Источник: fold.it
Своей идеей игра обязана участникам Rosetta@home — пользователи, наблюдая за тем, чем занимается их компьютер (программа, запустившись, выводит на экран скринсейвер, отображающий процесс подборки формы целевого белка), регулярно ловили себя на желании вмешаться в процесс. Сегодня игроки в Foldit предсказали и придумали уже несколько белков: для того, чтобы присоединиться к этому развлечению, не нужно ничего, кроме желания.
Именно так, через игры, в задачу по фолдингу белка пришли разработчики из DeepMind. По крайней мере, Демис Хасабис рассказывает, что он вспомнил про Foldit, когда смотрел на то, как созданная его командой программа AlphaGo одерживает верх над Ли Седолем. После «решения» го, алгоритм DeepMind уже совладал с набором из 57-ми классических аркад и стратегией в реальном времени StarCraft 2.
Кремниевые начинают и выигрывают
AlphaFold заняла первое место в CASP сразу же. Через два года точность предсказаний AlphaFold2 — ставшая трансформером, в отличие от своей свёрточной предшественницы — в 2/3 случаев оказалась, в общем-то, неотличима от экспериментальных данных. Третья версия, выпущенная весной этого года, опирается также на диффузионные модели (пока что архитектуру и код AlphaFold3 корпорация Google предпочитает хранить в тайне) и занимается уже не только предсказанием третичной структуры белка, но и малых молекул, и белковых комплексов.
Но что в принципе сделали в DeepMind, ясно и без технических деталей. Бейкер с коллегами показали, что объединение людей в сеть, причем неспециалистов, на порядки ускоряет поиск частного решения фолдинга белка — конкретной формы, которую принимает цепочка аминокислот. Иными словами, что достаточно коллективной интуиции. В Deepmind разработали алгоритм машинного обучения, который вобрал в себя экспертизу тысяч специалистов, собравших за полвека данные о структуре белков в базе PDB, — и продолжил играть в ту же самую игру на интуицию, уже с нечеловеческой скоростью.
Однако невероятный прогресс, которого мы добились за 30 лет соревнований CASP, остается сугубо вычислительным. Принципы, о существовании которых в 1972 году говорил Кристиан Анфинсен, остаются в 2024 году с определенностью существующими, но всё еще не познанными. Чтобы сделать конкретное предсказание, алгоритму AlphaFold иногда требуются дни размышлений и многие ватты энергии. Белки справляются за считанные мгновения.
Если бы эту Нобелевскую премия давали не в среду, а в понедельник, она была бы Нобелевской премией по медицине, а не Нобелевской премией по физиологии. Потому что техническое достижение лауреатов принесло огромное количество добра, но добра прикладного. Фундаментальная задача, то есть задача из области познания, продолжает стоять перед наукой в полный рост. Найдём ли мы её решение, изучая искусственные нейросети, которые, кажется, как минимум представляют его лучше нас, или сделаем это совершенно независимо от успехов кремниевой интуиции — вопрос открытый.
